
- 产品描述
-
Amber—分子模拟软件包
Amber力场是一套生物分子模拟的分子力场。Amber分子模拟软件包,由AmberTools和Amber组成,Amber是一个多个程序的集合包,大约包含60多个程序,每个程序之间可以很好的相互协调。Amber实行高效的并行计算,可以很好的处理大分子、有机物分子、隐(显)式溶剂模型,在生物、化工领域有广泛的应用。
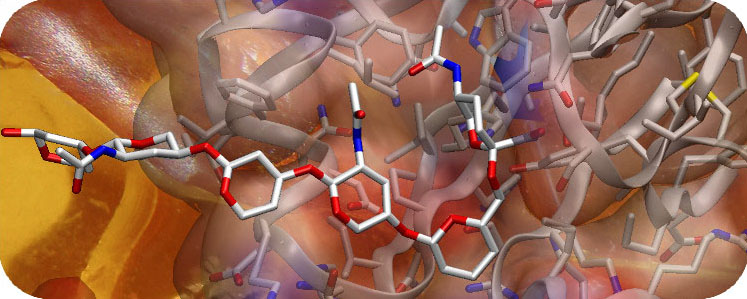
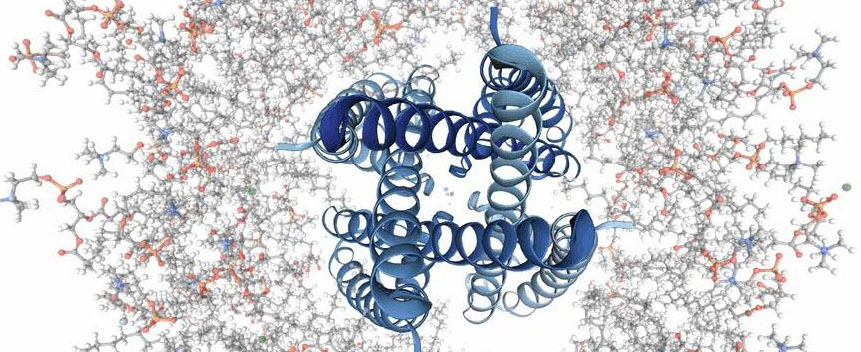
主要功能:
● 经典分子动力学模拟。
● 包括Quantum Mechanics/Molecular Mechanics(QM/MM)。
● 支持particle-mesh Ewald(PME)算法的显式溶剂模型,与generalized Born、Poisson-Boltzmann方法的隐式溶剂模型。
● 结构与轨迹文件分析。
● 用于生物分子模拟的力场(核酸、蛋白质、碳水化合物和有机物分子)。
● 支持并行动力学计算。
● 自由能计算(MM/PBSA等)。
主要程序
01
NAB/sff
该程序可以构建分子结构,使用generalized Born、Poisson-
Boltzmann或3D-RISM隐式溶剂模型运行MD或应用距离几何约束。02
Antechamber与MCPB
创建用于一般有机分子和金属中心的力场的程序。
03
tleap和parmed
Amber模拟的基本准备工具。
04
sqm
半经验和DFTB量子化学程序。
05
pbsa
对Poisson-Boltzmann模型进行数值解。
06
3D-RISM
求解积分方程模型以进行溶剂化。
07
sander
用于分子动力学模拟的主要程序。
08
gem.pmemd
使用高级力场的工具。
09
mdgx
主要通过参数拟合来突破Amber MD界限的程序。还包括可自定义的虚拟站点和显式的溶剂MD功能。
10
cpptraj与pytraj
分析轨迹结构和动力学的工具。
11
MMPBSA.py
基于能量的MD轨迹分析。
关键词:- Amber
- 分子模拟
- 有机物分子


上一页
解决方案
Solution
联系方式
公司地址:北京市朝阳区安慧里四区15号
中国五矿大厦419室
电话:010-64951848
传真:010-58603969
销售部:sales@tech-box.com.cn
泰科博思二维码
