
【精品课程!!!】DMol3模块之反应及分子动力学案例讲解专题班
在线报名 >
课程特色:
三合一学习:由计算老司机孙老师亲自授课,基础理论-软件操作-数据分析三个环节融为一体,通过对文献案例的拆解,将理论学习融入到计算操作中,不仅能输入能操作,而且能理解参数背后的设置逻辑。
计算模拟大局观:选择的前提是认知,即只有了解软件的基础理论方能建立起选择软件与方法的能力。实战中,既能充分利用不同软件的优势,又能有效规避特定方法的局限性,实现活学活用。孙老师长期一线科研、对多个软件的长期使用经验以及大量审稿经历,能有效帮助学员建立起计算模拟大局观。
学习的系统性:任何学习是有成本的,尤其是时间成本,因此计算模拟的学习一定要讲究系统性,实现螺旋式积累,与之相对的是碎片式学习,导致单一既能的盲目套用。在大局观建立后,孙老师通过精心设计小而巧的案例,将多达24个DMol3的计算技巧融入实际操作与数据分析中,这样不仅基础知识更清晰,而且能迅速将实际问题归入到上述同类案例,精准选择方法和计算指标,实现高效率计算。
选择Dmol3的四个理由:
A. 周期与非周期都支持:
DMol3几乎是为数不多的能同时支持周期与非周期计算的密度泛函软件;
B. 速度快:
由于采用数值基组,其计算速度非常快,而且能非常方便地实现局域轨道与电荷分析,可以轻松实现多达数百原子的理论计算;
C. 功能全:
目前DMol3已经包含各种泛函、溶剂校正、全电子基组支持、相对论效应等,同时支持激发态和各种光谱计算。
D. 分析方便:
DMol3作为Materials Studio的核心模块之一,能非常方便的利用可视化窗口实现数据分析,同时与其它MS模块无缝衔接,作图可以直接贴入论文。
讲师简介:
孙成华 教授
博士毕业于中国科学院金属研究所,导师为中国科学院院士成会明(《国家科学评论》编委)和中国科学院外籍院士、澳大利亚昆士兰大学逯高清(Max Lu,现为英国萨里大学校长,《国家科学评论》创刊副主编和顾问编委),主要研究领域为能源转化与存储,清洁环境及计算物理与化学。已先后在Nature,Nature Materials,Journal of American Chemical Society,ACS Nano,Angew. Chem. Int.Ed.,.Advanced Materials等国际知名刊物发表论文320多篇,他引22000多次,单篇最高引用超过4000余次,H因子66。先后在瑞典皇家理工学院、普林斯顿大学和哈佛大学以访问博士后学习,后正式加入澳大利亚莫纳什大学化学系,2017年加入斯威本科技大学,终身教职。
课程需知:
课程时间:9月24-25日(直播)
课程设计时长:共计12学时,24个技巧
课程费用:2480元
优惠:报名前15名,立减480元
1. 微信对公支付
2. 转账汇款
名称:北京泰科博思科技有限公司
银行:中国建设银行北三环支行营业部
账号:11001021200053001224
行号:105100005043
* 注意!!! 请将付款凭证及开票信息(需备注:专票or普票)发送至我司邮箱,info@tech-box.com.cn; 收到款后我们会根据您的要求为您开具发票。
报名方式:
1.点击上方"在线报名"提交报名信息
2.关注泰科科技微信公众号,对话框回复:报名,即可获得报名链接。
3.报名时请务必备注:Dmol3 专题班
4.若报名有困难,请联系房女士;info@tech-box.com.cn,电话:010-64951848,协助报名。
5. 我们会提前1~3天将具体上课链接发送至您邮箱,请注意查收。
6. 我们还设置专门的课程讨论群,欢迎进群交流。
适用学员:
课程面向零基础学员,有专门一节介绍密度泛函、软件窗口和各种功能,案例通用性强,均能使用笔记本在windows下点击完成(没有linux命令和脚本担忧)。
注意事项:
1.请参课人员自备软件(MS软件版本:8.0及以上,2020及以上最佳)
2.算例运行所需硬件条件:处理器:英特尔至强E5-1620或者等同性能
内存:8G(当前4000-5000元主流笔记本电脑即可满足)
3.课程观看目前仅支持Windows平台,暂不支持MAC OS系统
北京泰科博思科技有限公司是BIOVIA Materials Studio官方指定代理商,有关软件下详情或者技术支持请咨询北京泰科。
课程表(第一天):
上午 — 理论学习:(8-11)
1.DMol3模块与功能介绍
1.1 MS模块与DMol3的基本输入
1.2 DMol3对非周期/周期结构的支持
1.3 数值基组与独特的高效计算
1.4 赝势与泛函如何选择
本节需要建立的能力:
对MS和DMol3有快速了解,对数值基组、泛函、赝势有深入理解;进阶:极化/弥散基组特性、全电子基组;
2. 结构优化
2.1 晶胞优化 - 对称、元胞、约束优化
2.2 坐标优化 – 等同原子、坐标约束
2.3 表面优化 – 表面参数与面矢量
2.4 表面吸附优化 – 吸附构型设置
本节需要建立的能力:
理解结构优化判据;识别主要应力来源;理解初始构型扫描;建立梯度优化概念; 能解决收敛性问题;
3. 频率、能量与自由能
3.1 频率计算与虚频问题
3.2 总能量与零点能校正
3.3 自由能与熵的计算
3.4 反应自由能数据处理
本节需要建立的能力:
热力学量与换算; 电子能量与晶格振动能; 零点能; 振动频率与虚频消除; 化学反应完整热力学量处理;
上午 — 案例操作:(11-12)
4. Cu团簇优化与CO吸附
4.1 Cu4团簇优化
4.2 CO小分子优化
4.3 CO吸附构型扫描
4.4 频率与热力学量计算
本节需要建立的能力:
理解DMol3处理非周期结构的能力与必要性;团簇构型扫描;小分子吸附构型扫描;热力学量处理;
晚上 — 化学反应计算:(7-9)
5. 完整案例:CO2催化加氢
5.1 CO2加氢机理
5.2 初末态设置
5.3 过渡态搜索设置
5.4 过渡态频率验证
6.1 实际练习(20分钟)
6.2 过渡态虚频可视化与确认
6.3 虚频消除技巧
6.4 过渡态数据分析(能垒溯源)
本节需要建立的能力:
初末态原子对应问题;中间态插点与检查;过渡态精修;虚频查看与消除;虚频矢量可视化;
课程表(第二天):
上午 — 晶格动力学:(8-10)
7.晶格振动、分子光谱与声子谱
7.1 晶格振动基础知识
7.2 红外与拉曼光谱计算
7.3 声子谱计算
7.4 声子谱分析
本节需要建立的能力:
对MS和DMol3有快速了解,对数值基组、泛函、赝势有深入理解;
进阶:极化/弥散基组特性、全电子基组;
8.晶格振动实际操作
8.1 晶格振动的胞选择
8.2 红外、拉曼光谱数据可视化
8.3 固定原子的振动分析
8.4 声子谱K点设置与衍生量
本节需要建立的能力:
理解结构优化判据;识别主要应力来源;理解初始构型扫描;建立梯度优化概念; 能解决收敛性问题;
上午 — 分子动力学:(8-10)
9.分子动力学理论介绍
9.1 第一原理分子动力学基础
9.2 系综、热浴以及统计量
9.3 分子动力学典型设置
9.4 分子动力学数据分析
本节需要建立的能力:
热力学量与换算; 电子能量与晶格振动能; 零点能; 振动频率与虚频消除; 化学反应完整热力学量处理;
10.分子动力学实际操作(电池中的Li迁移)
10.1 MD任务提交和参数设置
10.2 动力学输出数据作图
10.3 扩散系数计算
10.4 轨迹分析初步
本节需要建立的能力:
理解DMol3处理非周期结构的能力与必要性;团簇构型扫描;小分子吸附构型扫描;热力学量处理;
晚上 — 电子结构分析:(7-9)
11.完整案例:单原子Fe催化N2活化
11.1 基底表面能带与态密度
11.2 Fe单原子投影态密度与电荷
11.3 孤立小分子N2的分子轨道
11.4 小分子吸附的参考态
11.5 小分子吸附后的投影态密度分析
11.6 局部电荷与差分分析
11.7 COHP计算分析
11.8 小分子与基底轨道作用分析
本节需要建立的能力:
建立电子结构分析的框架概念;会能带、态密度、局域电荷、分子轨道(含可视化)、差分电荷、COHP计算;拔高:轨道分析;态密度投影与电荷分析融合;
Dmol3精彩案例:
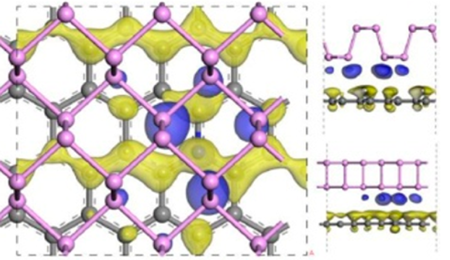
1. 石墨烯与磷烯异质结:
异质结是最常见的一种界面模型,对理论计算而言异质结非常容易因为体系过大而难以承受。这篇JACS展示DMol3计算石墨烯/磷烯异质结,清晰给出界面电荷转移(如下图),并用来解释这种电荷转移对催化性能的影响。
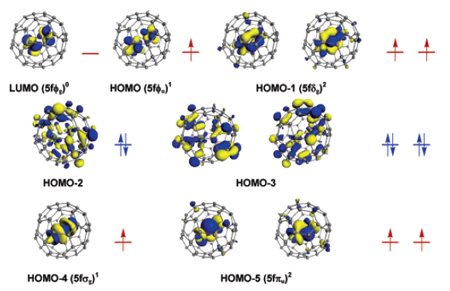
2. 稀土团簇中的磁耦合:
磁性体系,尤其是稀土磁性计算,往往需要测试不同磁序,进一步获取磁耦合信息。这个过程中需确保所用方法能区分不同磁序的细微差别,并对轨道占据有很好的描述,而周期结构往往因为存在自作用导致潜在风险。DMol3能极好支持团簇计算,因此处理带电团簇具有天然优势,同时其局域原子基组配合相对论赝势,使得处理稀土团簇非常方便(如U2的键合,JACS2007,下图),多篇JACS充分展示DMol这种能力。
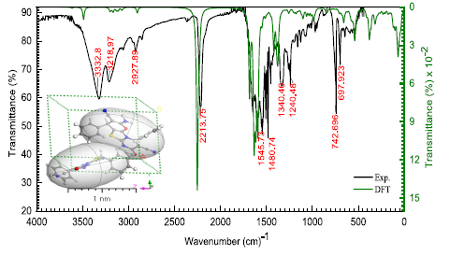
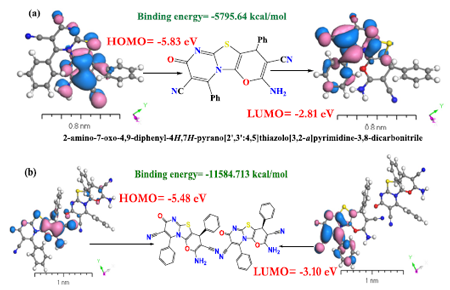
3. 全面的光谱计算与电子结构分析:
DMol3作为为数不多的软件,能全面支持各种电子结构分析,从基态到激发态均能计算(下图展示了含时密度泛函的光谱计算)。该工作通过分子间作用分析,获得了稳健的结构基础,然后分别对静电势、分子轨道以及激发态计算获得了发光机理的深入理解。这种统一软件下同时涵盖基态/激发态计算,并能方便进行数据可视化分析成为DMol3研究复杂分子体系的多种光谱成为有力工具。
关于北京泰科:
北京泰科博思科技有限公司(Beijing Tech-Box S&T Co. Ltd.)成立于2007年,是国内领先的分子模拟及虚拟仿真综合解决方案提供商。
北京泰科博思科技有限公司与国际领先的模拟软件厂商、开发团队深入合作,为高校、科研院所和企业在材料、化工、药物、生命科学、环境、人工智能及数据挖掘、虚拟仿真教学等领域提供专业的整体解决方案。用户根据需要在我们的平台上高效的进行各种模拟实验,指导实际的生产设计。
北京泰科博思科技有限公司拥有一支一流的技术服务团队和资深的专家咨询团队,以客户真正需求出发,服务客户,为客户创造价值。我们秉承“职业、敬业、担当、拼搏、合作”的企业精神,致力于用国际领先的软件产品和专业全面的技术支持服务,成为客户可信赖的合作伙伴。
Materials,Studio,DMol3

热门课程
11月28日 I 蒸汽压预测模型、活度系数模型、高通量计算问题、VLE相图中的EOS修正、Sigma文件输出方法等精彩内容等你来
2024-11-18 14:16
泰科云讲堂 I COSMOquick在共晶及低共融溶剂筛选中应用
本次讲座,将系统地介绍共晶筛选的多种方法、理论设计实现高效筛选、理论与现实筛选相结合的方法应用及展望、以及理论设计的COSMOquick软件的综合介绍等。
2024-04-18 10:57
本次云讲堂,北京泰科科技邀请来自北京化工大学材料科学与工程学院李豪祥博士和北京泰科技术总监郑宏博士,就分子模拟在高分子及橡胶材料领域的应用做相关的主题分享。
2024-04-08 15:09
【重磅课程!!!】基于Forcite模块攻破交联反应的模拟专题培训
主讲老师:月只蓝博士。课程提供独家脚本:专门适用于环氧+胺、环氧+酸酐、聚氨酯、交联成膜体系的Perl脚本;本课程中的脚本从未公开、全网唯一。
2023-09-05 14:24
【精品课程!!!】DMol3模块之反应及分子动力学案例讲解专题班
北京泰科科技推出,计算老司机孙老师亲自授课,基础理论-软件操作-数据分析三个环节融为一体,通过对文献案例的拆解,将理论学习融入到计算操作中,不仅能输入能操作,而且能理解参数背后的设置逻辑。
2023-09-05 09:29
【免费在线讲座】加速低共融容溶剂筛选:深度解析COSMOlogic软件的特点与优势
BIOVIA COSMOlogic是在COSMO-RS理论基础上发展起来的适用于任何混合物体系的流体热力学性质预测软件。
2023-08-24 15:06
联系方式
公司地址:北京市朝阳区安慧里四区15号
中国五矿大厦419室
电话:010-64951848
传真:010-58603969
销售部:sales@tech-box.com.cn
泰科博思二维码